

¿Es solo ‘intolerancia a la proteína’? Diferenciando y detectando la fenilcetonuria (PKU) más allá de la dieta
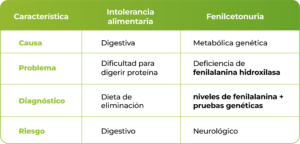
Cuando un niño presenta dificultades para tolerar ciertos alimentos ricos en proteínas, es común que surja la duda de si se trata simplemente de una sensibilidad alimentaria. Sin embargo, en algunos casos el problema puede estar relacionado con una enfermedad genética hereditaria conocida como fenilcetonuria PKU, un trastorno metabólico que afecta el metabolismo de los aminoácidos y que requiere diagnóstico y manejo especializado.
Confundir la PKU con una simple “intolerancia a la proteína” puede retrasar la intervención temprana y aumentar el riesgo de complicaciones neurológicas. Por ello, comprender cómo se diferencia esta enfermedad, cómo se detecta y qué papel juegan los niveles de fenilalanina en sangre es fundamental para profesionales de la salud y familias.
PKU vs intolerancia a la proteína: no son lo mismo
¿Qué es la fenilcetonuria (PKU)?
La fenilcetonuria PKU es un trastorno metabólico causado por una deficiencia enzimática que impide que el organismo procese adecuadamente el aminoácido fenilalanina. Esta alteración se debe a mutaciones en el gen PAH, responsable de producir la enzima fenilalanina hidroxilasa, que normalmente convierte la fenilalanina en tirosina.
Cuando la fenilalanina hidroxilasa no funciona correctamente, la fenilalanina se acumula en el organismo. Esta acumulación eleva los niveles de Phe y provoca un aumento de los niveles de fenilalanina en sangre, lo que puede dañar progresivamente las células nerviosas y afectar el desarrollo neurológico.
Las mutaciones genéticas en el gen PAH son la causa más común de este trastorno. La mutación del gen PAH altera la función de la enzima, generando una mutación de PAH que conduce al cuadro clínico característico de la enfermedad.
Sin tratamiento, los pacientes pueden desarrollar discapacidad intelectual, trastornos neurológicos y problemas del desarrollo, lo que históricamente llevó a clasificar la PKU como una causa prevenible de discapacidad mental.
Más que una intolerancia: una enfermedad metabólica
A diferencia de las intolerancias alimentarias comunes, la PKU es un síndrome clínico metabólico que afecta procesos bioquímicos esenciales del organismo. El problema no radica en la digestión de proteínas, sino en el metabolismo de los aminoácidos, específicamente en la incapacidad de transformar la fenilalanina.
Por esta razón, la PKU se considera una condición dentro de la genética médica, donde la alteración de un gen específico provoca una cascada de efectos metabólicos.
Los niveles de fenilalanina, deben mantenerse dentro de una concentración terapéutica segura para evitar daño neurológico. Si estos niveles se elevan de forma prolongada, pueden afectar el desarrollo cerebral, especialmente durante los primeros años de vida.
La importancia del tamiz neonatal
La detección temprana de la PKU se realiza mediante pruebas de detección incluidas en el tamiz neonatal, un programa de salud pública diseñado para identificar defectos congénitos metabólicos poco después del nacimiento.
Estas pruebas de detección sistemática utilizan análisis de sangre obtenidos mediante una pequeña muestra del talón del recién nacido. En el laboratorio se realiza la determinación de fenilalanina para evaluar los valores sanguíneos y detectar niveles elevados del aminoácido.
Si se identifican niveles de fenilalanina elevados, se realizan pruebas en sangre adicionales para confirmar el diagnóstico bioquímico de PKU. El seguimiento incluye el monitoreo de fenilalanina sérica y el control periódico de los niveles de Phe para ajustar el tratamiento
Confirmación diagnóstica y genética
Una vez detectada una alteración en los niveles de fenilalanina, el diagnóstico se confirma mediante pruebas genéticas que identifican mutaciones genéticas en el gen PAH.
Este proceso forma parte de la práctica de la genética médica y puede incluir la participación de un asesor genético, quien ayuda a interpretar los resultados y explicar los patrones de herencia a la familia.
El asesoramiento genético también es importante para futuras decisiones reproductivas, ya que la PKU es una condición hereditaria. En algunos casos, se pueden realizar estudios prenatales o análisis del ácido amniótico fenilalanina durante el embarazo.
Tratamiento: mucho más que evitar proteínas
El manejo de la fenilcetonuria se basa principalmente en el control dietético para mantener los niveles de fenilalanina dentro de rangos seguros.
La base del tratamiento es una dieta baja en fenilalanina, que implica una restricción dietética de alimentos ricos en proteínas. Esto incluye limitar ciertos alimentos sólidos como carnes, lácteos o huevos.
Para garantizar una nutrición adecuada, los pacientes utilizan alimentos médicos formulados para esta enfermedad, como:
- fórmula sin fenilalanina
- fórmula especial para PKU
- preparados nutricionales de fines médicos especiales
Estas fórmulas aportan proteínas sin fenilalanina, así como vitamina B12, suplementos minerales y otros nutrientes necesarios para el crecimiento.
El plan dietético suele estructurarse como un plan de comidas para la PKU o dieta de fenilcetonuria, que se adapta a la edad del paciente y se supervisa por un dietista metabólico bajo supervisión médica.
Seguimiento y control bioquímico
El manejo de la PKU requiere monitoreo continuo mediante análisis de sangre periódicos para evaluar los niveles de Phe.
Este control bioquímico permite ajustar la dieta especial, modificar la restricción dietética y mantener los niveles de fenilalanina en sangre dentro del rango recomendado.
El seguimiento debe realizarse en coordinación con profesionales de la salud especializados en enfermedades metabólicas, incluyendo pediatras, genetistas y nutricionistas clínicos.
PKU materna y riesgos durante el embarazo
Un aspecto crítico del manejo de la enfermedad es la Fenilcetonuria materna. Las mujeres con PKU que presentan niveles de fenilalanina elevados durante el embarazo pueden exponer al feto a concentraciones tóxicas del aminoácido.
Esto aumenta el riesgo de defectos congénitos, retraso del crecimiento y alteraciones neurológicas. Por ello, el control estricto de los niveles de Phe antes y durante la gestación es esencial.
El seguimiento incluye ecografías prenatales, monitoreo metabólico y apoyo de un asesor genético para reducir riesgos.
Detectar la PKU a tiempo cambia el pronóstico
La fenilcetonuria PKU demuestra cómo una enfermedad metabólica puede controlarse eficazmente cuando se detecta temprano. Gracias al tamiz neonatal, las pruebas de detección y el monitoreo de los niveles de fenilalanina, es posible prevenir complicaciones neurológicas y permitir un desarrollo saludable.
Reconocer que no se trata de una simple intolerancia alimentaria, sino de una condición metabólica genética que requiere diagnóstico y tratamiento especializado, es el primer paso para mejorar la calidad de vida de quienes viven con PKU.
Referencias bibliográficas
American College of Medical Genetics and Genomics. (2014). ACMG practice guideline for phenylalanine hydroxylase deficiency. Genetics in Medicine, 16(2), 188–200. https://doi.org/10.1038/gim.2013.157
Blau, N., van Spronsen, F. J., & Levy, H. L. (2010). Phenylketonuria. The Lancet, 376(9750), 1417–1427. https://doi.org/10.1016/S0140-6736(10)60961-0
Eunice Kennedy Shriver National Institute of Child Health and Human Development. (2023). Phenylketonuria (PKU). National Institutes of Health.
National PKU Alliance. (2022). Understanding PKU and dietary management. National PKU News.
Scriver, C. R. (2007). The PAH gene, phenylketonuria, and a paradigm shift. Human Mutation, 28(9), 831–845. https://doi.org/10.1002/humu.20526
Vockley, J., Andersson, H. C., Antshel, K. M., et al. (2014). Phenylalanine hydroxylase deficiency: Diagnosis and management guideline. Genetics in Medicine, 16(2), 188–200. https://doi.org/10.1038/gim.2013.157



